
 数据结构
数据结构 网络
网络 关系数据库管理系统 (RDBMS)
关系数据库管理系统 (RDBMS) 操作系统
操作系统 Java
Java iOS
iOS HTML
HTML CSS
CSS Android
Android Python
Python C语言编程
C语言编程 C++
C++ C#
C# MongoDB
MongoDB MySQL
MySQL Javascript
Javascript PHP
PHP中心核性肌病
中心核性肌病 (CNM) 是一组非常罕见的遗传性肌肉疾病。从轻微到严重的肌肉无力是几种疾病的标志。在极端情况下,症状可能在出生前出现,但也可能在任何时候出现,尽管成人发病并不常见。

什么是中心核性肌病?
中心核性肌病属于更广泛的先天性肌病家族的一部分。这些是遗传性肌肉功能障碍,通常在出生时或出生后不久就表现出来。根据医学文献,中心核性肌病指的是该病症的常染色体变异,而肌管肌病则描述了X连锁类型。由于X连锁肌管型CNM的症状往往比常染色体变异的症状更严重,因此做出这种区分至关重要。NORD在一份单独的论文中更详细地描述了X连锁肌管肌病。
肌肉活检通常会显示一种被称为中心核肌病 (CNM) 的异常,这是由于细胞核在肌纤维(肌细胞)内的位置不正确造成的。肌纤维的细胞核通常位于纤维的外部。在显微镜下检查肌肉活检组织,可以发现位于中心的细胞核,这是诊断CNM的关键特征。通过X染色体遗传的肌管肌病和几种常染色体类型(称为中心核性肌病)是CNM的众多类型中的两种。位于X和Y染色体以外的染色体上的基因被称为“常染色体”(性染色体)。虽然常染色体变异通常比XLMTM轻微,但在少数受影响的人群中,可能会出现类似于XLMTM中的严重后果。从轻度到重度不等的肌肉无力和肌张力减退(肌张力减退)是典型的表现。在最极端的情况下,可能会出现喂养问题和呼吸窘迫(呼吸困难)。参与进食和呼吸的肌肉受损会导致进食和呼吸困难。眼肌受累的表现范围很广。患者的症状可能从极轻微到婴儿期或幼儿期危及生命的后果不等。
中心核性肌病的症状有哪些?
不同类型的CNM以及个体可能会表现出差异很大的症状程度和严重程度。有些人可能只有轻度症状,而另一些人则可能出现严重的,有时甚至是致命的并发症。常染色体显性类型的CNM很少在儿童中致命,这与X连锁类型形成了鲜明对比。
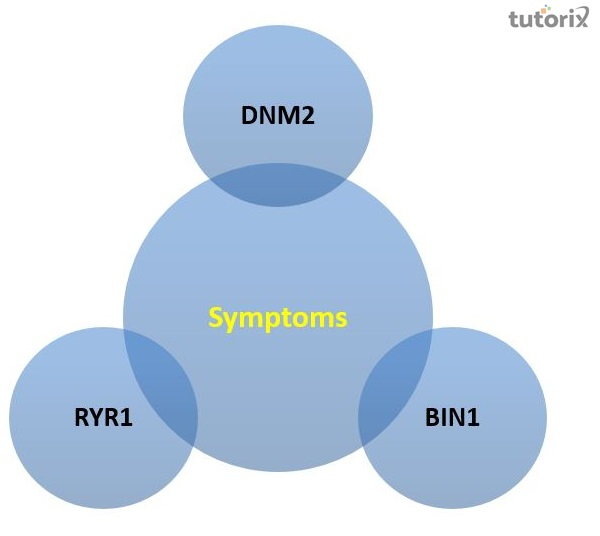
DNM2
DNM2基因的突变会在患有这种类型的CNM的人群中产生症状和体征。这种疾病可能从出生就存在,也可能在晚些时候出现。轻度形式的症状可能要到晚年才会出现,而另一些症状则可能在婴儿期或儿童期出现。
BIN1
这种类型的CNM以常染色体隐性方式遗传,由BIN1基因的突变引起。患有这种疾病的个体可能会有轻微到严重的症状,后者有时会在年轻时出现危及生命的并发症。尽管如此,目前只描述了少数几例与BIN1相关的CNM父母。因此,目前可能无法了解症状的全部范围。
RYR1
受影响者的特征性面部特征包括V形嘴。还记录了脊柱侧弯、反应缺失、眼肌麻痹和双侧眼睑下垂等症状。某些人可能会出现挛缩。一些出生时患有严重疾病的人在十几岁时取得了很大的进步,并且只有轻微的问题。
中心核性肌病的原因是什么?
蛋白质对于许多身体过程至关重要,并且是根据基因中的指令产生的。当基因发生突变时,产生的蛋白质可能功能失调或不存在。由于这取决于蛋白质的功能,许多不同的身体系统都可能受到影响。据认为,与中心核性肌病相关的基因编码的蛋白质在肌肉生长、维持和功能中起着至关重要的作用。这些蛋白质在体内的确切作用仍然是一个谜。不可能从父母双方遗传基因。遗传性疾病是由从父母双方遗传给定特征的DNA拷贝造成的。
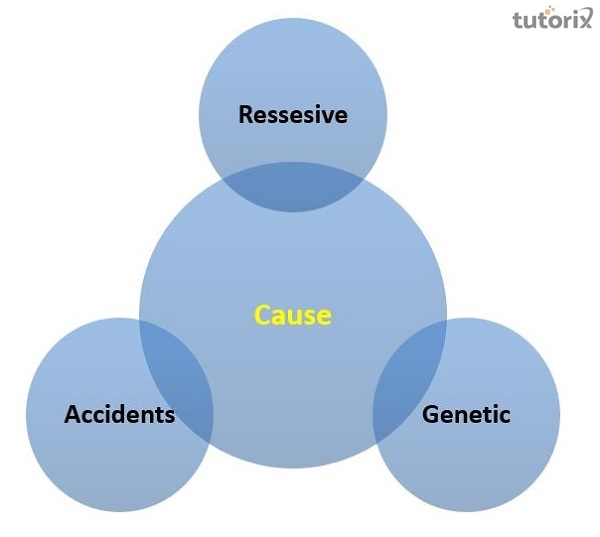
当一个人从父母双方都遗传了一个缺陷基因时,则称这种遗传疾病为隐性遗传。如果一个人遗传了一个功能正常的基因和一个该疾病的功能失调基因,则该人是该疾病的携带者。在两个携带者之间的每次怀孕中,父母一方将缺陷基因传递给孩子的风险为26%。孕妇分娩携带者孩子的几率为二分之一。孩子从父母双方都接受到功能正常的基因的几率是四分之一。
结论
像CNM这样的神经肌肉疾病是一项治疗挑战。然而,基因疗法正在被研究作为一种可能的治疗方案。鉴定CNM的致病基因为进一步研究这种治疗方案打开了大门。通过基因疗法用健康的基因替换患者受损的基因,可以恢复蛋白质合成并阻止疾病的进程。从理论上讲,这种治疗方法具有最高的成功概率,因为它涉及正常等位基因的永久修复,这可以在所有疾病部位产生活性蛋白。然而,在一种新的治疗方法可以作为一种可靠的替代治疗方法推广之前,仍然存在重大的技术障碍需要克服。

浏览量:70
