
 数据结构
数据结构 网络
网络 关系数据库管理系统 (RDBMS)
关系数据库管理系统 (RDBMS) 操作系统
操作系统 Java
Java iOS
iOS HTML
HTML CSS
CSS Android
Android Python
Python C语言编程
C语言编程 C++
C++ C#
C# MongoDB
MongoDB MySQL
MySQL Javascript
Javascript PHP
PHPDNA纯度评估
介绍
DNA纯度是指不含任何蛋白质、RNA或其他任何污染物的DNA。即使在DNA提取后,仍然会残留一些杂质,可以使用某些化学物质或酶将其去除。此步骤确保DNA的直接和下游应用的成功。
DNA纯化
使用各种物理和化学方法裂解细胞,以从细胞核中释放DNA。DNA提取后,细胞碎片以蛋白质的形式附着在其上,可以使用蛋白酶去除,或者直接从不纯的DNA样品中过滤掉。
进一步纯化是通过使用异丙醇或乙醇等醇类沉淀DNA来完成的。DNA溶于水,因此无法分离,因此使用乙醇搅拌DNA并使用无菌移液管将其缠绕出来。此后,DNA悬浮在碱性溶液中并使用。
DNA纯化的重要性
纯化很重要,因为它确保提取了研究工作所需的相同质粒或基因组DNA。
去除DNA中的碎片或污染物可以延长DNA的保存期,并降低研究工作中出现错误的可能性。
DNA纯度评估方法
一般使用三种方法来评估DNA的纯度:
吸光度法
荧光法
凝胶电泳
使用分光光度法的吸光度法
这是测试DNA纯度和产量的最可靠方法。吸光度法测定纯度所需的设备包括:具有紫外灯的分光光度计、对紫外光透明的比色皿以及待测DNA样品。
由于DNA在260nm波长处强烈吸收光线,因此吸光度读数取为260nm或A260。此波长产生的数值允许估计含有DNA溶液的浓度。
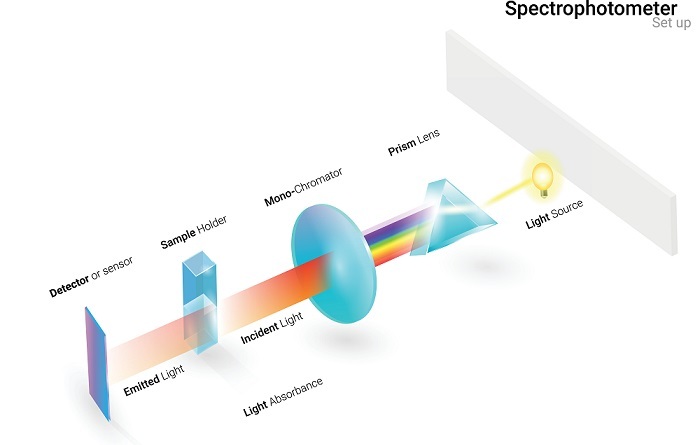
仪器中设置的线性范围为0.1到1,用于获得有用的数值。在320nm处测量DNA样品的浊度。
样品中DNA的浓度由以下公式给出:
DNA浓度(µg/ml) = (A260 - A320) × 稀释因子 × 50µg/ml
DNA的产量可以通过浓度和样品体积的乘积来测量。
DNA产量(µg) = DNA浓度 × 样品体积 (ml)
但是,这种计算存在局限性,不仅DNA,RNA也能在260nm处吸收紫外光。一些芳香族氨基酸,如酪氨酸、苯丙氨酸和色氨酸,也能在280nm处吸收紫外光,而胍在260nm处也有很高的吸收。由于所有这些原因,有时产量会被高估。
为了克服这一局限性,通过测试230nm到320nm的纯度来测试杂质的存在。最常用的纯度计算是通过取260nm与280nm的比率来完成的。纯DNA的值为1.7到2.0。低于1.7的值表示存在其他污染物。纯度计算公式如下:
DNA纯度 (A260/A280) = (A260读数 - A320读数) ÷ (A280读数 - A320读数)
纯DNA的首选值为大于1.5,小于该值表示浊度。
荧光法
与吸光度法相比,荧光法被认为更灵敏,可用于非常低浓度的DNA样品。
此方法使用荧光DNA结合染料和称为荧光计的仪器来检查DNA的纯度。与分光光度法相比,这些染料可提供更准确的结果。
一些常用的DNA结合荧光染料包括Pico Green、Quantiflour和Hoechst双苯并咪唑,它们特异性地结合到双链DNA上。
所使用的荧光计只有一个带有微孔板的试管,样品可以在PCR管、比色皿中读取,这使得它成为检测样品中DNA纯度和浓度的最便捷方法之一。
根据所选择的染料,设置激发和发射值,计算未知值的浓度并与已知值进行比较。
此方法的唯一局限性是荧光化合物的猝灭和光漂白效应会改变信号。
凝胶电泳
另一种用于测试浓度和纯度的方法是琼脂糖凝胶电泳。
此方法根据电荷和大小分离DNA。由于DNA带负电荷,它将向带正电的阳极移动,DNA分子越小,移动速度越快。
制备琼脂糖凝胶的百分比也决定了可以用其分离的尺寸范围。与以不同速率移动的RNA或蛋白质相比,形成的DNA条带是清晰的。
可以通过将DNA条带的强度与标准DNA进行比较来确定浓度,并使用嵌入染料溴化乙锭来观察DNA。
结论
在研究中,在将分离的DNA样品用于分析过程之前,质量指标显示其质量和可用性。这里的质量指标是DNA样品的完整性和纯度。纯度在很大程度上决定了研究的成功;因此,DNA样品应不含任何蛋白质等污染物。

327 次浏览
